
弗图约稿4 | 晚期糖化终产物及其不良反应:自噬
编者荐语:
以下文章来源于弗图医学,作者彗搏科技 李洋
摘要:
晚期糖化终产物(AGEs)积累在糖尿病和糖尿病并发症中所起的关键作用已得到广泛认可。AGEs会通过凋亡、炎症、蛋白质功能异常、线粒体功能异常和氧化应激等多种作用干扰几乎每个器官的正常功能。但是,目前尚未针对AGEs积累建立潜在的治疗策略。自噬是一种进化上保守的细胞过程,可通过降解和再循环系统维持细胞稳态。AGEs可以激活自噬信号传导,这可以作为针对AGEs诱导的问题的治疗策略。在这篇综述中,我们对AGEs的不利影响进行了概述,并提出了自噬可能是针对AGEs的有希望的靶向策略的观点。
关键词:晚期糖化终末产物(AGEs),AGEs‐RAGE轴,自噬,糖尿病,并发症,RAGE
01
介绍
糖尿病是一种以高血糖为特征的代谢综合征。由胰岛素抵抗或胰岛素缺乏引起的长期高血糖症是糖尿病发病机理中的关键因素。长期存在的高葡萄糖会上调不同的代谢途径,从而导致糖中毒或高血糖应激。这些代谢途径包括多元醇途径、糖酵解途径、己糖胺途径、蛋白激酶C(PKC)活化和晚期糖化终产物(AGEs)的形成。
高血糖症条件下,非酶糖化或AGEs的形成速率增加,并引起多种糖尿病并发症,例如视网膜病、白内障、神经病、肾病和动脉粥样硬化,以及伤口愈合延迟。非酶糖化(美拉德反应)是还原糖的羰基与蛋白质、脂质或核酸的游离氨基之间的反应。糖化改变蛋白质的结构和功能,从而对细胞过程产生不利影响。该反应开始于热力学上不稳定的席夫碱的形成,该席夫碱是高度可逆的。
这些Schiff碱随后被转化为Amadori产物,这些产品会进行一系列反应,最终形成不可逆的AGE。组织AGEs和循环AGEs的蓄积会导致不同病程的发展和进展,例如肾病、视网膜病、神经病和动脉粥样硬化。
因此,AGEs已受到世界各地研究人员的广泛关注,并且其作用已被广泛研究以寻找可对抗AGEs及其破坏性潜力的合适靶标。但是,针对AGEs的潜在靶标仍有待建立。
自噬是一个进化保守的过程,涉及降解或回收包括受损的衰老蛋白质和蛋白质聚集体在内的细胞成分。已知自噬对AGEs及其相关并发症起保护作用。通过在本领域进行适当的探索,可以将其开发为抗AGEs的潜在靶标,以进一步了解自噬在AGEs诱导的不良反应中的作用。因此,在这篇综述中,我们讨论了AGEs在各种系统中的影响以及自噬在减轻AGEs中的重要性。
1.1 方法
在审查的初始阶段,我们在电子数据库(例如PubMed,Google Scholar和Web of Science)中搜索了“AGEs和不良影响”之类的关键字,搜索结果产生了超过10万个结果。
在第二阶段,我们在相同的数据库中搜索了“自噬与AGEs”,并在Pubmed中找到了182篇文章,在Web of Science中找到了491篇文章,并在Google Scholar中找到了18,000篇文章。文章的进一步选择基于对适当标题和摘要的分析。最终数据收集是通过分析与本文相关的所选文章的全文来完成的。
1.2 晚期的糖化终产物
这些大分子中非还原糖和游离氨基之间的非酶促反应导致AGEs的形成。AGEs的形成过程会随着衰老和高血糖症而加速。AGEs与晚期糖化终产物(RAGE)受体的相互作用与大血管和微血管并发症的发生有关。
由于AGEs非常稳定,这些化合物会在组织中蓄积并破坏细胞功能,导致不同的病理生理状况,主要是糖尿病并发症,心血管疾病(CVDs),阿尔茨海默氏病和癌症。
2
AGEs的形成
AGEs的形成是一个复杂的多步非酶过程,称为美拉德(Maillard)反应,由Louis Camille Maillard于1912年首次报导. Maillard反应中涉及的化学反应由Hodge于1953年首先描述。
非酶过程可分为三个不同阶段:早期、中期和后期。该过程从还原糖的羰基与蛋白质,核酸和脂质上的胺残基之间的反应开始。此处形成的第一个产物是称为席夫碱的不稳定化合物。然后,该不稳定的席夫碱进行重排以形成更稳定的产物,称为Amadori产物(也称为早期糖化产物)(图1)。
在中间阶段,通过一系列反应、重排和脱水,这些Amadori产品会片段化为高反应性的二羰基化合物,例如甲基乙二醛(MG),乙二醛(GO)或脱氧葡萄糖酮(1-deoxyglucosone 和3 -脱氧葡萄糖苷)。二羰基化合物的积累会导致一种称为“羰基应力”的情况。羰基化合物可攻击蛋白质的赖氨酸,组氨酸,精氨酸或半胱氨酸残基。在后期,这些二羰基化合物会进一步与细胞成分发生氧化,脱水和环化反应,形成高度不可逆的褐色化合物,称为AGEs。
2.1 AGEs形成的其它途径
AGEs形成的其他途径包括氧化应激介导的葡萄糖氧化和脂质的过氧化导致的二羰基衍生物形成,二羰基衍生物可能导致AGEs形成。而多元醇途径是AGEs形成的另一种途径。
在此途径中,醛糖还原酶将葡萄糖转化为山梨糖醇,然后通过山梨糖醇脱氢酶将其转化为果糖,而果糖代谢产物是有助于AGEs形成的二羰基衍生物
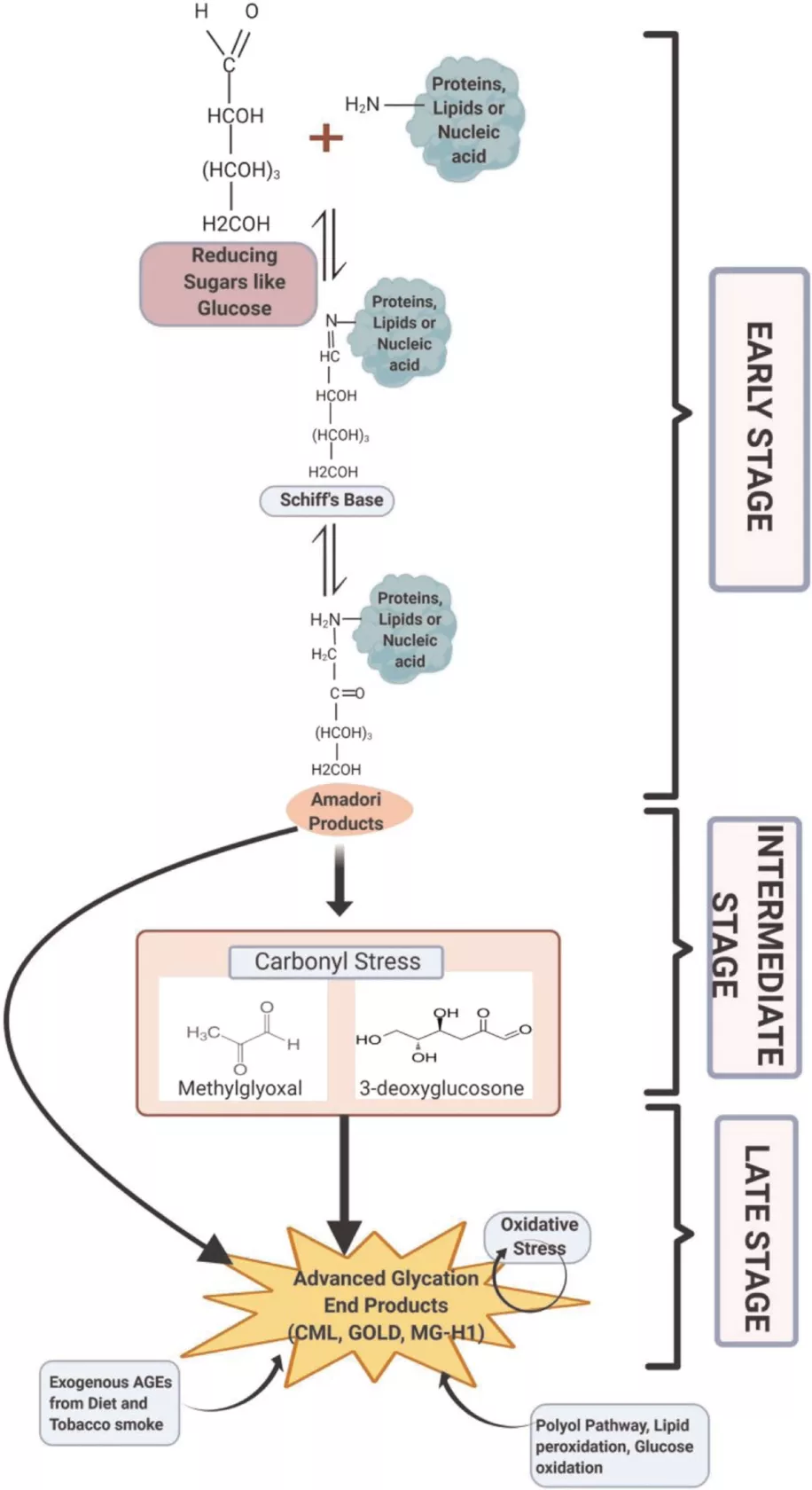
图1. AGEs的形成。蛋白质,脂质或核酸通过还原糖来糖化。 该过程涉及三个阶段,早期,中间和后期。非酶反应始于不稳定的席夫碱的形成,然后是Amadori产物,最后是不可逆的AGEs。氧化应激和AGEs在AGEs形成过程中遵循恶性循环。甚至饮食,烟草烟雾和各种内源性途径(如多元醇途径)也有助于AGEs库。AGEs,即advanced glycation end products,意为晚期糖化终产物。
3
外源性AGEs源
3.1 烟草
关于吸烟者中AGEs来源的信息很少。基于香烟的有毒反应性糖化产物,也称为糖毒素,到达肺泡,然后转移到肺实质细胞或血流中,在其中与血管壁蛋白或血清中的蛋白发生反应。在肺实质细胞中,糖毒素也可能与核酸反应以诱导突变。
另一项研究报告说,与其他来源(如葡萄糖)形成的AGEs可能需要几天到几周的时间才能诱导AGEs形成,而烟草中的糖毒素仅需几小时即可诱导AGEs形成。烟草中的糖毒素还预示着AGEs在血浆LDL和眼睛晶状体蛋白上的积累。
3.2 食物
食物中AGEs的存在是研究中最关注的领域之一,因为它有助于体内大量AGEs的产生。食物诱人的风味,气味和质地来自于加工食物过程中产生的AGEs。甚至未经烹煮的动物源食品也含有一定量的AGEs,随着烧烤,油炸和烧烤等进一步加工的增加,AGEs也会增加。
N?-羧甲基赖氨酸(CML)、戊糖苷、甲基乙二醛-赖氨酸二聚体(MOLD)和吡咯烷是食品中常见的AGEs。这些AGEs还只是少数,我们的饮食中还有许多其他AGEs分子需要鉴定。因此,用以分析食品中的这些分子的数据库仍然是一个更大的研究领域。
AGEs对我们身体的吸收可以通过简单的扩散或通过像肠道寡肽转运体(PEPT1)这样的肽转运蛋白来进行。摄入的AGE中只有约10%被吸收并分布到组织中,并且由于AGEs的交联键抵抗酶或酸水解,超过70%的AGEs不能吸收。
在肝脏,肾脏,肺,心脏和脾脏中发现了约60%的被吸收AGEs,显示了这些化合物的全身分布。吸收的AGE中有三分之一是通过肾脏排泄的。在较高的热量和较低的水分含量的烹饪条件下,AGEs的含量较高,而通过添加酸性溶液(如柠檬汁或醋)以及低温下更多的水分烹制的食品所含的AGEs相对较少。
3.3 AGEs的病理生理作用
3.4 与生命系统糖化相关的主要蛋白质
3.4.1 |胰岛素
胰岛素是一种肽激素,包含两条多肽链A(21个氨基酸残基)和B(30个氨基酸残基)。胰岛素通过刺激肌肉和脂肪组织(AT)吸收葡萄糖、抑制肝糖异生并刺激肝脏中糖原的合成来调节血糖浓度。胰岛素结构或浓度的任何差异都会导致其功能障碍。
作为具有51个氨基酸的肽激素,胰岛素还可以在B链的NH2末端苯丙氨酸(Phe1)处糖化。糖化胰岛素会失去控制血糖水平的能力。据报道,脂肪生成和葡萄糖氧化受损是胰岛素糖化的结果。最终,这导致了T2DM和相关的病理生理学(包括CVDs)的发展。已经证明,在糖尿病控制不佳的受试者中,循环糖化胰岛素的浓度显着增加。二糖化胰岛素在刺激葡萄糖摄取和糖原生成方面不如正常胰岛素有效。
据报道,大约10%的胰岛素糖化发生在胰岛中,与其他蛋白质糖化不同,胰岛素糖化发生在数小时而不是数周之内,这种糖化了的胰岛素会被β细胞储存和分泌。这些发现还指出了胰岛β细胞功能障碍和糖毒性之间的关系,β细胞功能障碍会导致T2DM中的胰岛素抵抗。糖化胰岛素显着性的评估可以使我们找到新的治疗途径。
据报道,糖化胰岛素对人脐静脉内皮细胞(HUVEC)和星形胶质细胞具有细胞毒性。它还会诱导活性氧(ROS)的产生和细胞凋亡,并可能增加可能导致神经退行性疾病的血脑屏障模型的通透性。在高血糖状态下,胰岛素糖化可能会加重人胰岛淀粉样多肽(hIAPP)聚集体的毒性,最终导致带来糖尿病状态的进展的β细胞死亡,从而揭示T2DM中的恶性循环。
3.4.2 血红蛋白(Hb)
3.4.3 |白蛋白
3.4.4 |胶原
胶原蛋白是一种在细胞外基质中发现的三螺旋结构蛋白。胶原蛋白中存在大量赖氨酸,精氨酸和羟基赖氨酸残基,使其更易于糖化和形成交联。蛋白质中基于AGEs的交联形成会导致组织的理化特性发生变化,并且大多数情况下会导致组织的变硬。
心肌和动脉僵硬是心血管系统中最令人担忧的问题,并且据信AGEs交联或长寿蛋白(如弹性蛋白和胶原蛋白)的糖化在其中起重要作用。糖化胶原蛋白的粘附与转化生长因子-β2(TGF-β2)调节的信号通路的激活有关,这反过来刺激依赖细胞信号转导分子3(Smad3)的α11整合素的表达,这与成纤维细胞形成和肿瘤变硬有关。
可溶性AGE直接参与胶原的交联。胶原原纤维的交联涉及糖尿病并发症和CVD。胶原蛋白无处不在,胶原蛋白的修饰会主要以丧失弹性的方式严重影响视网膜,皮肤,肌腱和韧带等组织。
3.5 AGEs诱导细胞反应
AGEs可能通过许多细胞内信号传导途径(例如JAK / STAT、活化B细胞的核因子κ轻链增强子(NF-κB)、PI-3K / Akt和MAPK / ERK途径)引起其反应。这些信号通路触发了多种导致AGEs的病理生理作用的转录因子的激活。
PI-3K / Akt和MAPK / ERK信号响应的激活导致NF-κB的激活,从而导致ROS的产生、炎症和细胞凋亡。JAK / STAT途径与多种自身免疫性疾病和细胞增殖有关。所有这些细胞效应主要受RAGE激活控制。
3.6 晚期糖化终产物的受体(RAGE)
RAGE是属于免疫球蛋白超家族的多配体受体。RAGE可以识别各种缺乏序列相似性的配体,因此被认为是模式识别受体。RAGE是由404个氨基酸残基组成的蛋白质,分子量约为55kDa。预计具有一个V(可变)和两个C(恒定)Ig结构域作为细胞外结构域,其后是穿膜区和具有小于50个氨基酸残基的短细胞质结构域。
RAGE在所有组织中广泛表达,包括肺,心脏,肝脏,骨骼肌和肾脏。除了肺中RAGE的表达更强以外,大多数组织的RAGE的基础表达都很低,特别是在肺泡上皮细胞中。还有一些研究报道了它们在先天和适应性免疫细胞中的表达。
全长RAGE:
可溶性RAGE:
sRAGE的两种形式是:
显性负RAGE(DN‐RAGE):
ΔN RAGE:
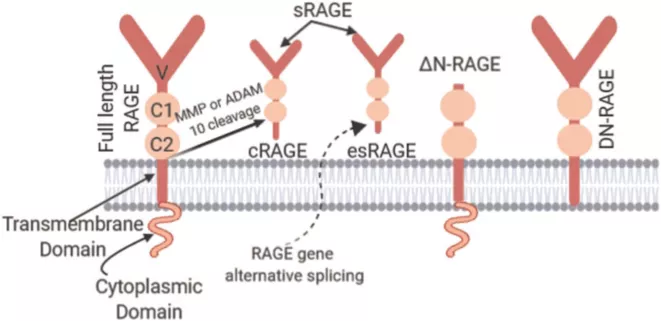 图2. RAGE的同工型。全长RAGE(具有细胞外结构域,跨膜结构域和细胞质结构域)参与信号转导。可溶性RAGE有两种形式:esRAGE(全长RAGE的剪接变体)和cRAGE(被细胞外蛋白酶切割的全长RAGE)。可溶性RAGE仅由细胞外结构域组成。ΔN-RAGE在V结构域中缺乏配体结合,而DN-RAGE缺乏细胞质结构域,两者均无法进行信号转导。cRAGE,晚期糖化终产物的裂解受体;DN-RAGE,晚期糖化终产物的显性负性受体;esRAGE,晚期糖化终产物的内源性分泌受体;RAGE,receptors for advanced glycation end products,晚期糖化终产物的受体。
图2. RAGE的同工型。全长RAGE(具有细胞外结构域,跨膜结构域和细胞质结构域)参与信号转导。可溶性RAGE有两种形式:esRAGE(全长RAGE的剪接变体)和cRAGE(被细胞外蛋白酶切割的全长RAGE)。可溶性RAGE仅由细胞外结构域组成。ΔN-RAGE在V结构域中缺乏配体结合,而DN-RAGE缺乏细胞质结构域,两者均无法进行信号转导。cRAGE,晚期糖化终产物的裂解受体;DN-RAGE,晚期糖化终产物的显性负性受体;esRAGE,晚期糖化终产物的内源性分泌受体;RAGE,receptors for advanced glycation end products,晚期糖化终产物的受体。
3.7 AGE对RAGE的激活
AGEs只是众多结合RAGE并激活细胞信号传导级联的配体中的一个。所有这些配体都具有相似的多β片层结构,从而使RAGE能够识别它们。AGEs-RAGE信号级联反应与许多疾病有关。特别是糖尿病及其相关并发症受AGEs-RAGE信号的影响很大(图3)。
AGEs-RAGE相互作用表明血管内皮生长因子(VEGF)和肿瘤坏死因子-α(TNF-α)的上调会降低血管屏障特性并增加DN的通透性。激活的信号级联直接影响细胞功能和代谢。损害的主要过程是由炎症和氧化应激引起的。在RAGE配体相互作用后激活的细胞信号传导途径包括JAK / STAT途径、MAPK / ERK途径、Src / RhoA途径、PI3K / Akt途径以及PKC和NADPH氧化酶的活化。
这些复杂信号级联的激活通过ROS的产生和转录因子(主要是NF-κB,AP-1和Egr-1)的激活而起作用。NF-κB的激活刺激各种炎症反应分子的表达和分泌,如环氧合酶2(COX-2)、白介素6(IL-6)、血管细胞粘附分子(VCAM)、细胞间粘附分子(ICAM)和TNF -α,从而调节AGEs-RAGE介导的炎症途径。
NF-κB激活还介导可诱导的一氧化氮合酶和NADPH氧化酶表达,从而导致过氧亚硝酸盐(ONOO-)的产生和氧化应激。AGEs / RAGE轴也有助于内质网(ER)应激,而NF-κB介导的ROS生成反过来会损害线粒体功能。RAGE激活还显示降低sirtuin 1(SIRT1)表达,从而导致线粒体生物发生受到损伤。
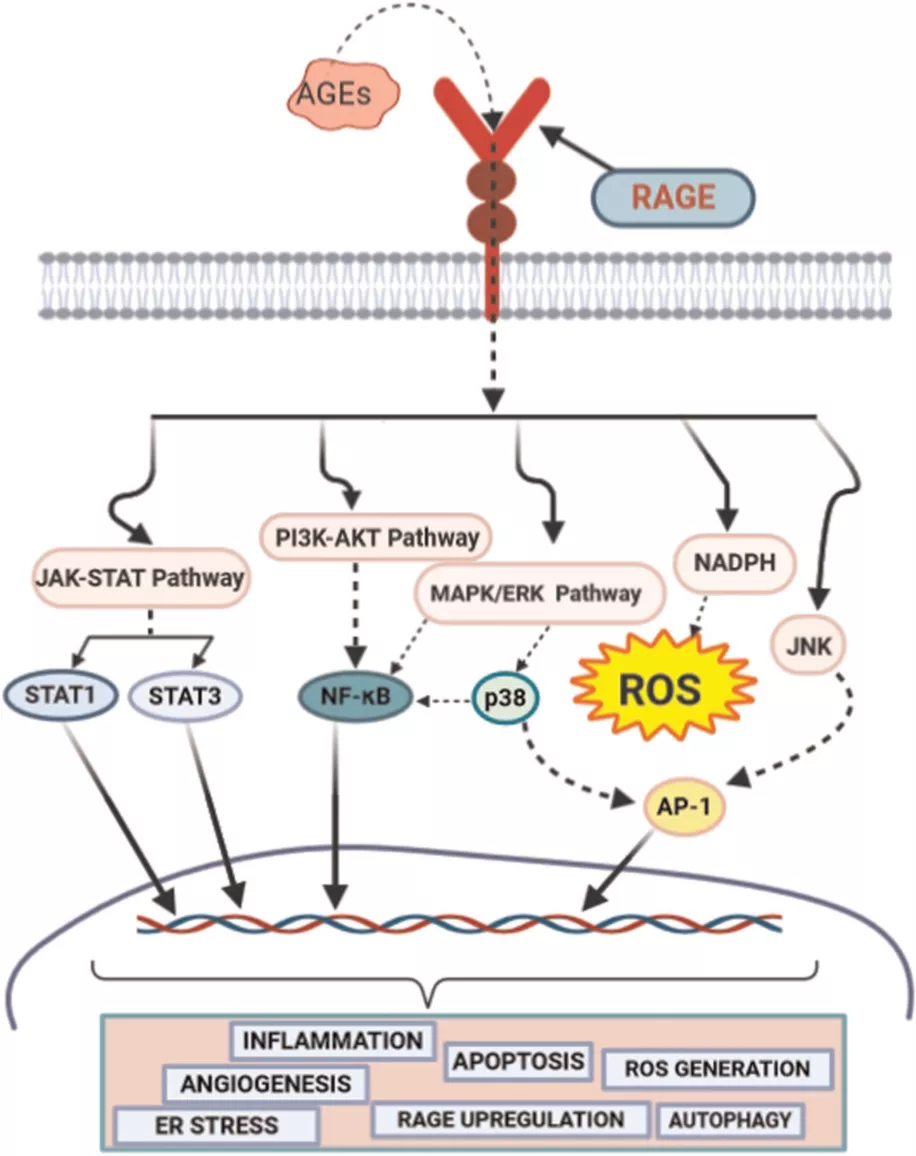
图3. AGEs-RAGE轴信令。AGEs-RAGE相互作用激活了不同的信号传导途径(JAK-STAT,PI3K-AKT和MAP / ERK途径)和信号传导分子。因此,触发各种转录因子的激活会带来核易位,并进一步引起炎症、RAGE表达上调、自噬、细胞凋亡、ER应激等。ER,内质网;ERK,细胞外信号调节激酶;JAK,Janus激酶;PI3K,磷酸肌醇3-激酶;RAGE,晚期糖化终产物的受体;STAT,信号转导子和转录激活子。
3.8 AGEs和氧化应激
氧化应激可以定义为防御系统中自由基和抗氧化剂来源之间的不平衡。氧化应激是许多疾病(包括糖尿病和心血管疾病)的病理生理学中公认的参与者。AGEs和氧化应激之间存在复杂的关系,因为两者的产生是相互联系的。
AGEs的积累会产生氧化应激,而氧化应激的生成会加快AGEs的生成和积累。AGEs-RAGE相互作用诱导ROS生成和炎症途径的级联反应。据报道,具有抗氧化特性的酶的糖化可提高ROS的产生并引起细胞的氧化损伤。已知抗氧化剂可防止AGEs诱导引起的NF-κB、TGF-β1活化和细胞死亡。
AGEs的形成减少了抗氧化酶(如超氧化物歧化酶(SOD)和过氧化氢酶)的活性,并诱导了ROS的产生。据报道,参与MG解毒的乙二醛酶I(GLO-1)的过度表达会降低糖尿病大鼠的GO,MG和AGEs水平,并降低ROS的产生,表明AGEs与氧化应激之间存在相关性。
氧化应激还会提高丙二醛(MDA)的水平,丙二醛可形成与兔脑中的AGE相对应的被称为ALE的蛋白质加合物。AGEs激活依赖Nrf2的抗氧化基因NADPH:醌氧化还原酶1(NQO1)和血红素加氧酶-1(HO-1),为糖尿病的内源性氧化应激提供保护。AGEs与氧化应激之间的复杂关系仍需探索以进一步了解相关并发症。RAGE激活与急性全身注射脂多糖引起的氧化性肝损害有关。
3.9 AGEs,RAGE和内质网
ER是真核细胞中最大的膜细胞器。ER在细胞中具有多种功能,例如蛋白质合成、蛋白质折叠和运输、脂质和类固醇合成、钙存储和碳水化合物代谢。为此,ER配备了独特的蛋白质和酶来维持体内平衡环境,这些环境很容易被导致ER应激的细胞环境变化所破坏。
蛋白质合成和折叠过程中的任何混乱都会在管腔中积聚错误折叠或未折叠的蛋白质,从而激活一系列信号反应以清除该混乱,这个机制被称为未折叠蛋白质反应(UPR)。UPR由三个主要传感器组成,即蛋白激酶RNA样内质网激酶(PERK),肌醇需求酶(IRE1)和激活转录因子6(ATF6)。
如前所述,AGEs通过修饰和灭活酶、蛋白质、脂质和核酸而参与细胞的活动的总干扰,因此也会引起ER应激。鼠足细胞的AGEs暴露会导致通过ER应激途径诱导的细胞凋亡。导致ER应激的AGEs积累似乎在肾脏、大脑和心脏的低氧损伤中也有作用,表明了它们在发病机理中的中心作用。
AGEs对骨关节炎的参与也与RAGE的激活的ER应激信号通路相关,ER应激信号通路激活会导致人软骨细胞中COX-2表达和NF-κB通路激活。AGEs前体3-DG被认为与ER应力诱导的caspase-3活化有关,而与成纤维细胞中的RAGE活化无关。据报道AGEs通过ER应激通过内皮细胞的凋亡参与内皮功能障碍。ER应激的产生和自噬似乎也参与了由AGEs引起的肾小球膜细胞凋亡。
AGEs通过增加淀粉样蛋白β和ROS的产生在阿尔茨海默氏病中起作用,粉样蛋白β和ROS通过削弱Sirt-1的神经保护能力诱发ER应激引起神经元细胞死亡。通过以时间和浓度依赖的方式增加ER应激标志物(如GRP78,ATF4,CHOP和拼接的XBP1)的水平,钙稳态的失调和ER应激的启动会导致添加了AGEs的髓核(NP)细胞的凋亡。
所有这些研究清楚地表明,AGEs诱导的ER应激会导致炎症和细胞凋亡,从而在几种疾病中对组织造成严重损害。
3.10 AGEs,RAGE和线粒体功能障碍
线粒体长期以来与多种疾病相关,例如阿尔茨海默氏病、胰岛素抵抗、T2DM、癌症、心血CVD、不育症、非酒精性脂肪肝疾病(NAFLD)、衰老等。许多研究人员已经将线粒体与人类疾病的这种关系与由于氧化损伤导致的线粒体功能障碍联系起来。线粒体被认为是AGEs引起的氧化应激的重要来源。
AGEs可通过RAGE途径引起线粒体功能障碍,从而引起线粒体形态的变化并破坏嵴和其他内部结构。用CML-牛血清白蛋白(BSA)处理β细胞会导致RAGE表达增加、线粒体膜电位去极化、ROS增加、线粒体DNA缺失、线粒体动力蛋白失衡以及ATP含量降低,表明线粒体严重受损可能导致线粒体自噬和胰岛素分泌受损。
心肌细胞添加AGEs会导致神经酰胺积聚并减少线粒体呼吸,揭示了了AGEs、线粒体和CVDs间的联系。当用AGEs处理时,人脐静脉内皮细胞(HUVEC)由于线粒体呼吸和糖酵解的减少而表现出能量缺乏,这导致细胞活力和增殖能力的抑制。添加AGEs还诱导成骨细胞凋亡,这和RAGE途径介导的线粒体异常有关。为了更好地了解线粒体功能障碍及其与代谢性疾病的关系,线粒体糖化及其后果仍有待进一步阐明。
3.11 AGEs和炎症
越来越多的证据表明,炎症在几乎所有疾病(例如糖尿病,CVD,癌症,炎症性肠病等)中都起着核心作用。由于AGEs的积累,氧化应激和炎症同时进入了恶性循环。如前所述,AGEs与RAGE的相互作用激活了包括炎症在内的多个细胞信号级联反应。
在与衰老相关的病理过程中,无菌炎症或无病原体的炎症是由危险相关分子模式(DAMPs)介导的。DAMPs可以与特定的受体(例如RAGE)相互作用以诱导无菌炎症,而AGEs就是可以与RAGE相互作用以诱导无菌炎症反应的无菌危险配体。
NF-κB在激活AGEs-RAGE轴下游的众多蛋白质方面起着主要的转录调节剂的作用。NF-κB的激活开启了几种促炎因子的分泌,例如IL-6,IL-1β和TNF-α。
在AGEs诱导的人脐静脉内皮细胞(HUVEC)中,细胞间黏附因子-1(ICAM-1)和血管细胞粘附分子-1(VCAM-1)的表达与其他促炎细胞因子(如IL-6,IL-1β和TNF-α)一起增加,表明AGEs诱导了炎性血管内皮功能障碍。
链脲佐菌素诱导的糖尿病小鼠喂食高AGEs会导致的血清TNF-α和IL-6含量升高,并对肾脏和心脏等器官造成严重损害。
3.12 AGEs和心脏病
AGEs诱导的胶原蛋白交联一直是一个令人关注的问题,因为它会导致舒张功能障碍。AGEs会由于肌内质网Ca2+ -ATP合酶泵和鱼尼丁受体等细胞内蛋白质的交联而干扰心肌细胞的松弛和收缩特性。
甚至由于载脂蛋白B100糖化导致的低密度脂蛋白摄取减少也与动脉粥样硬化有关。内皮细胞中的AGEs / RAGE轴激活ER应激、炎症和氧化应激等多种有害作用的信号途径,从而引发细胞骨架重排,进而导致细胞通透性增加。
同样,血管平滑肌细胞(VSMC)中的AGEs / RAGE信号转导会激活基质金属蛋白酶-2/9(MMP-2/9)、炎性细胞因子和ER应激途径,导致血管平滑肌增生和细胞外基质降解,以及自噬和溶酶体降解受损。
3.13 AGEs与肝脏疾病
多项研究表明AGEs与肝病有关,如肝纤维化、肝硬化和NASH。在高AGEs和高脂饮食喂养的小鼠中,NAFLD的发展与肝脏炎症有关。在高AGEs饮食的小鼠中,可以观察到参与NAFLD进程的四个枢纽基因(Cidea,Cidec,Fabp4和Plin4)的表达升高。RAGE的表达增加表明,AGEs / RAGE轴参与了NAFLD的肝损伤和炎症反应,提示AGEs / RAGE途径是要考虑的治疗策略。
多项研究表明,从患有肝病、纤维化和肝硬化的患者采集的样品中,CML的浓度增加了。在慢性丙型肝炎患者中也已证明自噬参与了肝星状细胞的活化,而后者通过AGEs / RAGE轴参与了肝纤维化。甚至AGEs / RAGE在肝细胞癌(HCC)中的作用也广受关注。RAGE在HCC进展中的增殖、侵袭和血管生成等不同阶段中起的作用也引起了该领域的强烈兴趣。
3.14 AGEs和肾脏疾病
肾近端小管细胞通过吸收肾小球滤过液中的AGEs并使其分解代谢,从而在AGEs代谢中起主要作用。这些AGEs可以在近端肾小管上皮细胞(PTEC)的细胞表面与RAGE相互作用,并激活级联的细胞内信号通路。
AGE / RAGE轴诱导依赖于ER应激的p21信号传导,导致PTEC提前衰老。糖尿病期间由MG的形成和积累引起的二羰基应激及其通过GLO-1减少的解毒作用导致DN的发展。在成熟的足细胞中,AGEs激活Notch 1信号传导,这可能导致蛋白尿或肾小球疾病。
3.15 自噬在糖化中的意义
3.15.1 |自噬概述
3.15.2 |自噬的类型
3.17 自噬机制
通过鉴定酵母中近30种自噬相关基因(ATG),可以了解哺乳动物的自噬机制。该机制的基础是保守的,在很多哺乳动物中都找了和酵母菌对应的的ATGs。
3.18 自噬体形成
吞噬泡是一个小的杯状结构,是自噬体前体或自噬体前结构(PAS)的起源。自噬体形成是由雷帕霉素复合物1机制性靶标(mTORC1)的抑制引发的。饥饿、ER应激、氧化应激和缺氧或任何传染性病原体侵袭等应激条件都会引发mTORC1的抑制。自噬体形成发生的第一步是组装unc‐51样自噬激活激酶(ULK1)复合物,该复合物在吞噬泡上,包含ULK1、Atg13、FIP200和Atg101四个亚基。
在饱食状态下,mTORC1抑制ULK1;因此,抑制mTORC1可使ULK1复合物激活。自噬体形成受到乙酰化和蛋白质磷酸化的控制和协调。ULK1一旦被激活,就会磷酸化并激活beclin1。自噬体形成启动之后是自噬成核阶段,在此另一个含有包括Beclin1在内的几种蛋白质的复合体被激活。该复合体称为Beclin1 / III类磷脂酰肌醇-3-激酶(PI3k)复合物。
复合物包括Beclin1、白血病B细胞淋巴瘤因子-2(BCL-2)、ambra1、Atg14、抗紫外线辐射相关的肿瘤抑制基因(UVRAG)、内啡肽B1、Vps15、PI3K和液泡蛋白分选蛋白34 (Vps34)。
Beclin 1复合物激活PI3K,并将磷酸肌醇二磷酸酯(PIP2)转化为磷酸肌醇三磷酸酯(PIP3)(图4)。PIP3将WD重复阈磷酸肌醇相互作用蛋白(WIPI蛋白)募集到前自噬体膜上。P62(泛素货物结合蛋白)和NBR1伴随WIPI而来,充当选择性靶底物的受体。
下一步是前自噬体膜的伸长,它由微管相关蛋白1A / 1B-轻链3(LC3)蛋白和Atg5-Atg12-Atg16L1复合物协调,该复合物有助于磷脂酰乙醇胺的缀合,从而使LC3-II易位延伸形成成熟的自噬体。LC3偶联系统包括将LC3蛋白转化为LC3-I,然后再转化为LC3-II,参与偶联系统的关键参与者是Atg4,Atg3,Atg7和Atg10。
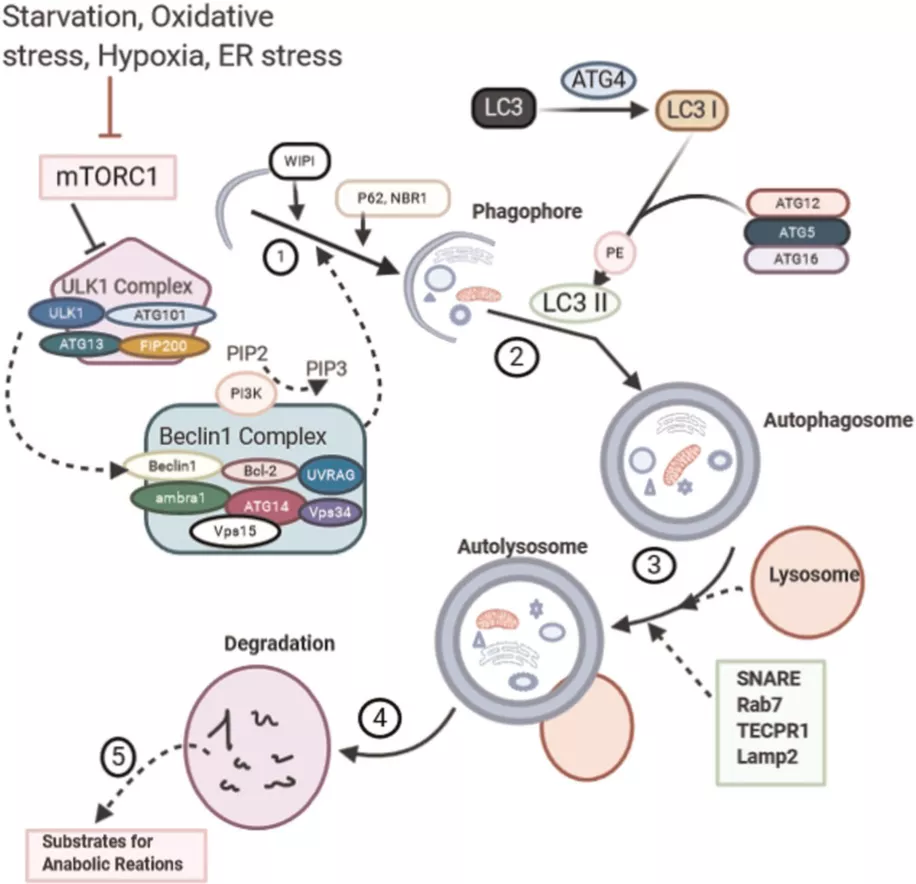
图4. 自噬信号转导。信号是由AMPK依赖的mTORC1抑制作用引发的,mTORC1的抑制作用依次刺激ULK1复合分子激活beclin复合分子,然后激活PIP3。(1)成核,PIP3将WIPI和p62(泛素结合蛋白)和NBR1蛋白募集到常染色体前,用于选择性底物的成核。(2)吞噬细胞膨胀并与目标底物形成封闭结构的成熟过程称为自噬体。(3)融合,在此阶段,自噬体在衔接子和束缚蛋白的帮助下与溶酶体融合形成自溶体。(4)回收,此阶段涉及货物底物的降解和(5)回收成代谢物和营养物质。mTORC1,雷帕霉素复合物1的机制靶标;ULK1,unc-51像自噬激活激酶; WIPI,WD重复阈磷酸肌醇相互作用蛋白
3.19 自噬体与溶酶体融合
一旦自噬体形成,它就与溶酶体融合形成自噬溶酶体。自噬体的成熟受到溶酶体膜蛋白(LAMP2)和内体标记蛋白Rab-7和SNARE蛋白的调控。小型GTP酶(例如RAB7A,RAB2A,ARL84A / B)负责控制自噬体-溶酶体融合,衔接子和束缚蛋白(HOPS,EPG-5,ATG14L,TECPR1,GRASP55,BRUCE,RUFY4)将自噬体与溶酶体结合在一起。
自噬体与溶酶体之间的融合是通过SNARE蛋白完成的。一旦融合,自噬酶体中的内容物就被溶酶体酶降解为氨基酸,脂质或核苷酸,并通过渗透酶转移到细胞质中。这些降解产物被细胞用于各种合成代谢反应,以维持其在应激条件下的可持续性。
3.20 糖尿病中的自噬
3.20.1 |胰腺β细胞的自噬
3.20.2 | 骨骼肌自噬
骨骼肌在胰岛素介导的葡萄糖摄取中起主要作用,因此是T2D中胰岛素抵抗的重要靶标。在骨骼肌中,运动,禁食和萎缩会刺激自噬。
据报道,小鼠自噬不足可导致GLUT4表达受损,葡萄糖敏感性/耐受性降低以及葡萄糖摄取减少。因此,骨骼肌自噬的损害与T2D中胰岛素抵抗的进展有关。
3.20.3 | 脂肪组织自噬
胰岛素介导的葡萄糖摄取的另一个主要部位是AT。比较带有非糖尿病肥胖的T2D患者的脂肪组织的自噬和正常苗条个体的脂肪组织自噬,可以在T2D患者中观察到自噬的上调。
据报道自噬对于过氧化物酶体增殖物激活受体(PPAR-γ)介导的脂肪细胞分化和脂肪形成至关重要。Atg7基因敲除小鼠体型较瘦,并因更高的葡萄糖利用率,胰岛素敏感性和β-氧化作用而具有更高的代谢率。

3.20.4 |年龄和自噬
自噬是AGEs的有效清除途径,但是随着年龄的增长,这种途径会减弱。几项研究显示了在AGEs处理的细胞系中诱导出了细胞自噬。但是,该领域仍需积极探索以寻找其积极作用或消极作用的相关事实。
自噬作为细胞保护性清除途径降解不需要的或不再需要的细胞成分时,其过度活化可能导致自噬凋亡。关于AGEs诱导的自噬的保护作用有许多报道,而一些报道说AGEs诱导的自噬是糖尿病创伤愈合受损的原因。
然而,Han等综述了自噬在清除AGEs以修复糖尿病创伤方面的临床应用。然而据报道,在糖尿病性血管并发症中, FOXO1与AGEs诱导的内皮细胞自噬凋亡有关。AGEs诱导的自噬也通过ERK和Akt途径促进了动脉粥样硬化的病理发展(图5)。
有迹象表明自噬对AGEs诱导的凋亡具有保护作用。AGEs处理可增加源自肌腱的干细胞,软骨细胞和成骨细胞中LC3B / LC3A的比例。
基于MG的糖化应激通过激活多囊卵巢综合征卵巢中的自噬标记物而参与了SIRT1 / AMPK信号通路介导的小鼠卵巢功能障碍。
另一项研究表明,MG诱导的自噬对两种细胞保护蛋白,硫氧还蛋白1(trx1)和乙二醛酶2(GLO-2)的降解具有有害作用。AGEs诱导的VSMCs钙化通过HIF-1α/ PDK4诱导的自噬途径的激活而减弱。
但是,心肌细胞中的毒性AGEs抑制了LC3-II / LC3-I的表达,从而减弱了导致细胞死亡的自噬途径。Mei等指出了ROS / ERK途径在AGEs处理的人牙周膜细胞的自噬诱导中的作用。
也有报道说,RAF蛋白激酶和NF-κB参与了AGEs诱导的自噬的刺激,表明RAGE激活途径也参与其中。自噬在抑制肾脏衰老中的保护作用,以及RAGE / STAT5信号通路在抑制AGEs处理的肾小球细胞自噬的作用也得到了证实。sRAGE引发的STAT3依赖性自噬抑制作用已显示出对心脏缺血/再灌注(I / R)损伤的保护作用。
RAGE在胰腺癌自噬的调节中也起着关键作用,RAGE的抑制与自噬一起减轻了小鼠模型中的肿瘤生长和肿瘤发生。AGEs通过抑制与AMPK / mTOR信号通路相关的自噬来促进血管钙化。AGE-RAGE-自噬轴也与癌症进展有关。对暴露于AGEs的H9c2细胞进行β-胡萝卜素治疗可以逆转自噬的升高,建议将β-胡萝卜素作为AGEs引起的心脏功能障碍的一种保护措施。
AGEs抑制了巨噬细胞中的自噬通量,从而降低了金黄色葡萄球菌感染期间的细胞内杀菌能力。表皮AGEs与皮肤暗淡的外观有关,并且自噬的激活被认为是一种有效的治疗方法。据报道,卤代富乃酮通过连续自噬可抵抗AGEs诱导的H9c2细胞损伤。
AGEs处理的系膜细胞中自噬的抑制导致ROS的产生和凋亡细胞的死亡。成骨细胞中AGEs诱导的ROS生成和线粒体损伤被发现激活了针对细胞凋亡的自噬反应。在足细胞中,AGEs抑制TFEB的核易位并激活mTOR信号传导,从而抑制自噬体的形成和周转。
在人软骨细胞中AGEs在时间和浓度的背景下对自噬的双相作用也已经被证实。AGEs在短时间内低剂量诱导自噬,而长时间高剂量则抑制自噬。AGEs诱导增加HUVEC中p62的表达和LC3-II / I的比率。Verma和Manna证明了p53在不同AGEs处理的细胞中自噬和凋亡之间的细胞反应切换中的作用。
因此,关于自噬作为朋友或敌人的作用的清晰见解需要透彻理解和深入研究,以有效地针对糖尿病和衰老过程中的AGEs和AGEs引起的并发症靶向该途径。
3.20.5 | AGEs诱导CVD和自噬
几项研究报道自噬参与了不同的CVDs,如心肌病,动脉粥样硬化,I / R和心力衰竭。VSMC中AGEs-RAGE的激活会诱导ROS的产生、炎症、ER应激和缺氧,所有这些都会导致细胞增殖和迁移,从而导致动脉粥样硬化病变的发展。在心血管病理生理学中,自噬可能在不同条件下在不同细胞系中扮演朋友或敌人的角色。
自噬在局部缺血过程中扮演朋友的角色,而在再灌注过程中扮演敌人的角色。另一项研究证明了自噬的长期激活在心脏肥大发展为心力衰竭中的作用。RAGE / PI3K / Akt / mTOR途径可以激活AGEs诱导的心肌细胞中的自噬。
RAGE自噬轴参与心力衰竭被证明是治疗的新靶标,因为RAGE抑制作用减弱了心肌细胞的自噬死亡。MG诱导血管内皮生长因子受体2的自噬降解,并在糖尿病的血管生成受损中起主要作用。
因此,需要对该区域进行更多的探索,以更好地了解自噬在CVD中的细胞保护作用或细胞病变作用。药物开发的标准,无论是激活还是抑制,都将根据患者的病情而有所不同。
3.20.6 | AGEs诱导DN和自噬
自噬被认为是针对DN的新型治疗靶标。据报道,氨基胍是一种著名的抗糖化化合物,可以恢复糖尿病引起的肾溶酶体加工过程中的调节,这种调节是DN发生的初始事件。溶酶体组织蛋白酶活性的改变导致肾小管细胞蛋白降解受损,从而导致异常蛋白的积累,并因此导致糖尿病性肾肥大。
HK-2细胞的AGEs-BSA暴露会通过AGE / RAGE相互作用破坏自噬酶体途径,并导致异常的蛋白质积累。科学家们已经研究了几种针对DN的潜在目标分子,包括AGEs抑制剂氨基胍,但不幸的是尚未达到药物研发的最终阶段。然而,这种严重的糖尿病并发症的流行在全世界范围内正在增加。因此,迫切需要针对DN的新型治疗靶点,自噬是一个值得考虑的好方法。
3.21 |转化意义
如前所述,AGEs及其不良反应表明它们严重参与了各种病理生理状况。潜在的抗糖化剂的开发将对许多疾病的治疗有效。到目前为止,市场上没有可用的药物,研究人员正在寻找针对AGEs的潜在分子靶标。自噬研究领域是一个有希望的突破。
自噬的参与涉及多种疾病模型。更好地了解自噬如何参与AGEs引起的并发症,将成为有关糖尿病和相关疾病的新视角。用激活剂、抑制剂或基因操纵调节自噬途径可能是一种抗击疾病的有用方法。自噬在不同条件下的作用令人惊讶地矛盾。此外,自噬及其在多种疾病中的作用尚未得到探索。因此,它们正在拓宽药物开发领域的新视野。
4
结论
这篇综述的目的是帮助读者理解AGEs的各种不利影响以及与AGEs相关的信号通路和自噬之间的联系。自噬在维持细胞环境中的稳态方面起着关键作用。这篇综述中的大多数研究表明了自噬作为一种针对AGEs诱导的细胞损伤的保护措施的作用。将自噬作为抗AGEs和相关糖尿病并发症的防御工具是一个合理的选择。对于研究人员而言,重要的是了解并批判性评估AGE与自噬之间的联系。而为了开发自噬疗法,仍需要深入的研究来了解AGEs诱导自噬的基本生物学。
微信扫一扫
关注该公众号
- 线粒体与皮肤健康、衰老和疾病(上篇)
- 没有了!